Pôle de Chimie Calculatoire de l'UJF
Responsable scientifique : Serge Pérez, Centre de Recherches sur les Macromoélcules Végétales (CERMAV, UPR 5301) Tel: 04-76-03-76-30, Fax: 04-76-03-76-29; Mèl: perez@cermav.cnrs.fr
Responsable technique : Pierre Vatton, Laboratoire d’Etudes Dynamiques et Structurales de la Sélectivité (LEDSS, UMR 5616) Tel :04-76-51-48-02 , Fax: 04-76-51-43-82; Mèl: Pierre.Vatton@ujf-grenoble.fr
I. Introduction
Au sein de l’Université Joseph Fourier, une communauté scientifique répartie sur plusieurs unités de recherche et sur plusieurs sites, partage des préoccupations communes autour de la chimie ‘calculatoire’ qui regroupe la chimie quantique, la mécanique et la dynamique moléculaires, les méthodes de type Relations Structure-Activité (QSAR) et Relations Structures-Propriétés (QSPR). Les outils bio-informatiques et les consultations des bases de données sont des pratiques de plus en plus fréquentes, qui accompagnent les développements permanents qui résultent des séquençages des génomes.
L’expérience montre qu’entre les stations de travail et les gros serveurs de calcul des centres nationaux, il y a place pour une informatique intermédiaire et de proximité, qualifiée de méso-informatique, qui est capable de fournir localement une puissance de calcul importante, tout en maintenant la fonctionnalité et la souplesse des stations de travail. Cette souplesse est primordiale dès lors que l’on travaille avec des chercheurs ou des étudiants sur des thématiques nouvelles et non rodées. Cette souplesse est aussi fondamentale pour la réalisation de travaux qui requièrent plus de 48 heures de monopolisation de ressources importantes car il est évident qu’on ne peut affecter à un utilisateur, pendant trop longtemps, toutes les ressources d’une machine nationale. Par ailleurs, il convient de prendre en compte l’interactivité graphique que requièrent les logiciels de modélisation moléculaire et de disposer de ressources informatiques locales susceptibles de supporter des charges de travail importantes. Dans ce domaine, la forte demande d’interactivité de divers applicatifs de traitements des données expérimentales (cristallographie des protéines, molécules, complexes moléculaires,… , RMN multi-dimensionnelle, spectroscopies infra-rouge et Raman,….) nous obligent également à avoir localement une grande puissance de calcul. Les centres de calculs nationaux que les chimistes grenoblois utilisent largement (15000 heures par an en moyenne) sont réservés aux travaux planifiés qui auront été préparés et mis au point localement sur les machines locales de méso-informatique.
Le Pôle de Chimie Calculatoire de l'UJF ne pouvant se faire qu’en regroupant des acteurs de la recherche qui partagent les mêmes intérêts et les mêmes outils, il créera une synergie importante au sein de la communauté des chimistes grenoblois. Il permettra également de partager les coûts des logiciels d’application qui représentent une part de plus en plus importante dans les budgets de fonctionnement.
La présente demande est faite au nom de cinq unités de recherches qui au sein de l’Université Joseph Fourier à Grenoble, partage des besoins communs, immédiats ou futurs dans l’utilisation de machines de type méso-informatique. Il convient évidemment de souligner que cette demande est également faite dans un esprit qui permette l’intégration des unités et thématiques de recherches émergentes, tout en respectant les efforts souvent importants que peuvent consentir des unités de recherche, dans le bon fonctionnement de ce service commun..
II. Les unités de recherches concernées
:Le projet, présenté au nom de l’UFR de Chimie, implique de manière forte le laboratoire de Cristallographie (UPR 5031), le Laboratoire d’Etudes Dynamiques et Structurales de la Sélectivité (LEDSS, UMR 5616), le Centre de Recherches sur les Macromolécules Végétales (CERMAV, UPR 5301), le Laboratoire d’Electrochimie Organique et de Photochimie Rédox (LEOPR, UMR 5630), et le Département de Pharmacochimie Moléculaire (DPM, UMR 5063) de l’UFR de Pharmacie.
Figure 1 : les partenaires
L’ensemble de ces laboratoires recouvre une population des plus de trois cent chimistes, physico-chimiques, pharmaciens, biochimistes,… Au travers des actions de recherche et des projets des différentes équipes des laboratoires partenaires, peuvent être énoncés les principaux domaines de recherches et d’applications qui suivent : la chimie fine, (synthèse des produits naturels, catalyseurs, complexes récepteurs-substrats, extractants de radio-éléments, anti-cancéreux, anti-rejets, nutriments,..) les macro-molécules biologiques (protéines, oligo- et polysaccharides, oligonucléotides,…) et leurs conjugués, les matériaux… Les substrats étudiés au sein des différents programmes de recherche sont de nature multiple. Ils se caractérisent par une distribution de masse moléculaire couvrant plusieurs ordres de grandeurs (de quelques dizaines d’atomes, à plusieurs centaines, voire plusieurs milliers). Ces substrats peuvent se présenter soit sous forme gazeuse, liquide, gel, colloïdale, solide cristallin, solide amorphe,…
III. Projets Scientifiques Concernés.
Les potentialités scientifiques des unités de recherches partenaires de ce projet leur permettent de s’inscrire totalement dans la dynamique de création et de fédération d’un centre de méso-informatique du pôle Chimie de Grenoble. Au travers des réalisations récentes et surtout des projets scientifiques des chercheurs de ces unités, apparaissent des besoins de matériels informatiques et de logiciels, dont la mise en commun permettrait à cette communauté de conserver une position scientifique de pointe dans plusieurs domaines scientifiques. Les programmes scientifiques des différentes unités de recherche qui sont présentés de manière succincte, peuvent être regroupés au travers de quelques grandes catégories qui sont représentées schématiquement sur la Figure 2.
Figure 2 : Les axes scientifiques des différents partenaires
III.1 Les Méthodes Quantiques :
III.1.a. Réactivité
La réactivité représente un axe de recherches développées de manière importante sur le site de Grenoble, en particulier au Laboratoire d’Etudes Dynamiques et Structurales de la Sélectivité. La détermination des intermédiaires et états de transition nécessaires à la caractérisation des chemins réactionnels, requiert l’utilisation de l’ensemble des méthodes de la chimie quantique. Les méthodes post-Hartree telles que le niveau de calcul CCSD(T) permettent d’obtenir des résultats très fiables pour des systèmes à couches fermées, mais malheureusement de petite taille (5 à 10 atomes). Pour des systèmes de 30 à 40 atomes, le niveau de calcul MP2 permet d’obtenir une bonne estimation des grandeurs thermodynamiques et cinétiques. Pour des systèmes à couches ouvertes ou présentant des états quasi-dégénérés, il convient d’utiliser des méthodes multi-configurationnelles de type CASSCF, CASPT2, MRCI qui ont prouvé leur fiabilité. Les méthodes de type fonctionnelle de la densité (DFT) permettent d’inclure, à moindre coût, la corrélation électronique dans les optimisations de géométrie. Cette approche est particulièrement intéressante pour les systèmes organo-métalliques qui comportent des états proches en énergie et, souvent un nombre important d’électrons. De plus, la qualité des résultats des méthodes DFT hybride est souvent comparable à celle des niveaux de calculs comme CCSD(T) et de façon générale, elle est proche du niveau MP2.
Réactivité radicalaire : Les additions de radicaux libres sur les molécules non saturées représentent un moyen commode de créer de nouvelles liaisons. Ils jouent un rôle essentiel dans de très nombreux processus de polymérisation et interviennent dans de nombreux processus biologiques. Des études systématiques ont été entreprises, qui incluent les effets de solvants, sur l’aptitude des méthodes de la fonctionnelle de la densité, à traiter les problèmes de réactivité et de régio-sélectivité radicalaires. L’objectif de ce travail est double : (i) mieux appréhender les facteurs qui gouvernent la réactivité et l’orientation de ces réactions; (ii) élaborer un protocole fiable qui permette de traiter des ensembles atomiques importants, susceptibles de mimer ceux rencontrés dans des systèmes d’intérêt biologiques. Les réactions étudiées concernent l’addition du radical méthyle sur des éthènes diversement substitués et une meilleure compréhension de la régio-sélectivité.
Réactivité organométallique : La synthèse de cyclopentanones de façon régioséléctive, à partir d’un alcyne, d’un alcène et d’un complexe hexacarbonyldicobalt représente un exemple traité actuellement en utilisant la théorie de la fonctionnelle de la densité. Le but de cette étude est une meilleure compréhension du mécanisme de cette réaction en apportant un support théorique aux expérimentateurs dans leurs tentatives d’obtention de nouveaux ligands pour des complexes bicobalts, puis de rendre la réaction plus stéréosélective vis à vis de l’alcène et d’induire une énantiosélectivité.
Réactivité non radicalaire : L’étude des mécanismes de cyclo-addition (4 + 2; 3 + 2; 2 + 2; 3 + 3) constitue un domaine d'études particulièrement important qui est développé en partenariat avec des expérimentateurs. Les réactions modèles telles que l’addition du butadiène sur l’éthylène, le formaldéhyde et le thioformamide illustrent les travaux réalisés dans le cas des cycloadditions de type 4 + 2. Les autres types de cyclo-additions sont traités dans le cadre de la dimérisation de la sulfine H2CSO. Les effets de solvants sont également pris en compte dans la cadre : (i) de réarrangements sigmatropiques faisant intervenir un atome de soufre pour former des liaisons C-C avec C=CH2 dans le but d’obtenir des acides aminés optiquement actifs; (ii) de la réaction entre un aldéhyde avec de l’acide cyanhydrique et de l’ammoniac en présence d’eau (synthèse prébiotique d’a acides aminés; (iii) d’addition nucléophile sur le disulfure de carbone.
III.1.b. Interactions
La description des interactions inter-moléculaires peut être menée par deux méthodes qui sont souvent utilisées de façon complémentaire: l’approche supermoléculaire et la théorie des perturbations. L’approche supermoléculaire consiste simplement à calculer l’énergie d’interaction entre deux molécules, en soustrayant à l’énergie de la supermolécule, la somme des énergies des monomères qui la constituent. Une alternative à cette approche consiste à calculer directement l’énergie d’interaction en utilisant la Théorie de Perturbation avec Adaptation de Symétrie (SAPT), qui permet la décomposition de l’énergie d’interaction en termes physiquement interprétables par la physique classique (termes électrostatiques ou d’induction) ou par la physique quantique (termes d’échanges ou de dispersion). Cette méthode a aussi été développée pour les interactions à trois corps, c’est à dire à trois molécules. Parmi les nombreux domaines d’applications, ceux développés à Grenoble concernent la spectroscopie (trimère HCl(H2O) transitions spectroscopiques correspondant à un effet tunnel entre deux minima, énoncé des règles de sélection, schéma d’intensité des raies observables en spectroscopie de rotation), chimie ‘environnementale’ (agrégats HCL(H2O) : adsorption et réaction de HCL au niveau des calottes glaciaires sont responsables de la formation de Cl2, qui dans la stratosphère, forment les radicaux Cl* responsable de la destruction de la couche d’ozone), astrophysique (interaction entre l’ammoniac NH3 et l’hélium, reproduction théorique de spectres haute résolution).
III.1.c. Vers une meilleure description de la corrélation électronique
Parallèlement au traitement de molécules toujours plus importantes, une puissance toujours accrue des ordinateurs permettrait d’étudier les problèmes chimiques à un niveau de calcul plus fiable, i.e. en améliorant la description de la corrélation électronique. Si les chimistes quanticiens se sont toujours intéressés à la description des molécules (énergie, géométrie, répartition de charges, orbitales moléculaires, propriétés électroniques (moment dipolaire, polarisabilité, …) ils n’oublient pas que l’essence de la chimie a pour origine, et finalité, l’interaction entre molécules. L’interaction entre deux molécules est un préliminaire à toute réaction chimique. L’interaction d’une molécule avec le solvant peut influer de manière prépondérante sur sa réactivité. Les interactions inter ou intra moléculaires vont conduire à certaines conformations du complexe moléculaire, qui peuvent, à leur tour, déterminer la réactivité de ce dernier, comme c’est souvent le cas pour des molécules biologiques. Ces conformations, sont responsables de la structure des spectres haute résolution.
La description des interactions, quelle que soit la méthode utilisée reste un domaine délicat d’un point de vue théorique et très exigeant quant aux moyens informatiques requis pour une étude ad hoc.
III.2. Mécanique et Dynamique Moléculaires.
Les méthodes de mécanique et dynamique moléculaires offrent une alternative intéressante aux méthodes quantiques, dans la mesure où elles permettent l'étude de systèmes moléculaires et macromoléculaires complexes,
Comme les mouvements électroniques ne sont pas traités explicitement, les méthodes de dynamique moléculaire sont incapables de prédire la formation ou la rupture des liaisons pas plus que la géométrie et les propriétés de systèmes pour lesquels la délocalisation des électrons ou les interactions entre orbitales atomiques jouent un rôle important. Elles présentent toutefois des atouts indéniables par rapport aux méthodes quantiques: (i) elles sont rapides et ne nécessitent pas une puissance de calcul démesurée ce qui permet d'aborder l'étude de gros systèmes moléculaires; (ii) elles sont simples à mettre en œuvre; (iii) la géométrie et les énergies relatives obtenues sont souvent d'une qualité égale à celles fournies par les méthodes quantiques. Les résultats sont d'autant meilleurs que le système étudié est proche des structures qui ont servi à la paramétrisation.
On recense de nombreux logiciels construits autour de champs de force de complexité et de précision variables; certains sont de nature généraliste (MM3) ou plus appropriés à telle ou telle famille de molécules et macromolécules. Les meilleurs logiciels de mécanique/dynamique moléculaires, souvent accompagnés d'outils de construction et de visualisation, sont utilisés sur le site Grenoblois (Discover, Tripos, Charmm, Amber, Jumna, Cerius,…). Ils sont utilisés en fonction de la nature et la spécificité des substrats étudiés (ADN, protéines, polysaccharides, polymères, molécules et complexes moléculaires,…). Le domaine d'application de ces logiciels s'est considérablement élargi, au travers de l'utilisation d'algorithmes d'exploration de l’hyperespace conformationnel avec des techniques de plus en plus précises: cartes d'énergie, dynamique moléculaire, méthode de Monte Carlo, méthodes heuristiques,…
Dans certains domaines d'applications, ces études de mécanique moléculaire sont menées sur de molécules modèles, considérées comme fragments d'unités plus complexes telles que des polymères. Ces informations permettent de calculer les moyennes de grandeurs observables: rayon de giration, longueur de persistance, et d'identifier les bases moléculaires qui régissent les relations structures-propriétés.
La dynamique moléculaire est une simulation du mouvement des atomes et des molécules par calcul de leurs déplacements. Elle permet d’aborder l’étude de très gros systèmes et de prendre en compte l’aspect temporel des phénomènes. Cette technique est très utilisée pour simuler les propriétés des solides, des liquides et des gaz. Dans le cas de molécules d'intérêt biologique et/ou technologique, telles que le saccharose ou le tréhalose, la prise en considération explicite de la solvatation, sur des échelles de temps de plusieurs nanosecondes, a permis d'établir les bases moléculaires responsables de certaines propriétés spécifiques. A l'interface chimie-biologie, des programmes de recherche développés au LEDSS et au CERMAV étudient les conformations de macromolécules en prenant en compte de manière implicite ou explicite les molécules de solvant. La compréhension de mécanismes réactionnels de protéines, ou la dynamique de leurs interactions avec des substrats ou des analogues de substrats, représente un volet d'étude très développé sur le site grenoblois. Une telle avancée s’explique aussi par le rapprochement, de plus en plus marqué au cours de ces dernières années, entre chimistes théoriciens et chimistes expérimentateurs. Ce rapprochement sera d'autant plus effectif que les simulations permettront d'aborder l'étude de systèmes moléculaires plus importants, sur des durées de simulation plus proches de celles observées expérimentalement, et que les composantes électrostatiques seront prises en compte. Autant de contraintes exigeantes quant aux moyens informatiques requis pour une étude ad hoc.
III.3. Etat condensé
III.3.1. Matériaux moléculaires:
Les méthodes classiques de résolution structurale trouvent leurs limites dans l’étude des molécules organiques. Très performantes dans le cas où il y a redondance de données expérimentales par rapport à la complexité du modèle structural (rapport nombre d’observations / nombre de variables), elles deviennent inopérantes lorsque la condition de redondance n’est pas réalisable. La modélisation numérique, grâce à la dynamique moléculaire avec contraintes, permet d’intégrer des données expérimentales (diffraction, RMN, IR…). Cette potentialité a permis la détermination sur monocristaux de structures cristallographiques de molécules pharmaceutiques comme celles des anti-convulsifs de type aximino substitués.
Développées pour les études sur monocristal de molécules organiques, ces méthodes sont actuellement appliquées à la diffraction sur poudre et conduisent à des résultats très prometteurs, en particulier lorsque les échantillons sont micro-cristallisés et (ou) affectés de polymorphisme. Cette technique acquiert une grande efficacité lorsqu’elle est associée aux simulations numériques : recuit simulé, méthode de Monte-Carlo inverse, dynamique moléculaire avec contraintes et algorithmes génétiques.
La détermination " ab initio " d’une structure à partir de données de diffraction des rayons X synchrotron, sur poudre, permet par exemple le positionnement et/ou la connaissance de la conformation de groupements spécifiques dans une maille cristallographique de composés pharmaceutiques. L’ajustement final du modèle structural est alors réalisé par affinement des paramètres de torsion et de translation de la molécule. Des structures ont ainsi pu être déterminées à partir de données de diffraction sur poudres. Il faut ajouter à cela les calculs de quantification de phases sur poudres, liées au polymorphisme des molécules pharmaceutiques.
Dans le domaine de la science des matériaux, cristaux pour l’optique non linéaire, la recherche d’empilements simples de chromophores organiques ( ions, molécules) qui optimisent la susceptibilité non-linéaire du second ordre c (2) est toujours un défi. La plupart des structures connues, certes très efficaces, n’optimisent jamais le tenseur c (2). Le rôle de la modélisation dans cette ingénierie est de calculer les hyperpolarisabilités statiques b xyz des chromophores (MOPAC+PM3 Hamiltonien) en fonction de la symétrie de la molécule proche de celle rencontrée dans le cristal. Ensuite, la somme des bxyz moléculaires sur tous les sites dans le cristal conduit aux coefficients du tenseur c (2). En modifiant la paramétrisation (distribution de charges) du chromophore et son orientation dans le cristal, on modifie et on optimise le tenseur c 2) ; on peut donc reconnaître les empilements favorables.
Ainsi, par exemple, l'efficacité du cristal (P21) de chlorure de 2-amino-3-nitropyridinium, est expliquée par le caractère bi-dimensionnel des hyperpolarisabilités du chromophore cation (b xxx et b yxx) en présence de l'ion Cl-. On peut signaler aussi l'accroissement des non-linéarités dans les anions 4-nitrophénolates par transfert de proton (accroissement de b ingénierie de sels organiques par transfert de proton.
Enfin, on citera le tetra-(4-methoxy-phenylphosphonium) iodide qui contient un cation chromophore tétraédrique octupolaire: évaluation des b au niveau du cation tétraédrique pour comprendre l'efficacité du cristal.
Le polymorphisme, très souvent présent dans ces cristaux, produit des matériaux non centrosymétriques très efficaces dont les structures ne sont pas identifiables par manque de données aux rayons X. Ici aussi la modélisation, recherche de structures optimales avec une symétrie propre, peut aider à la découverte de nouvelles phases. Le calcul de morphologie cristalline, énergies suivant les directions d’empilements des molécules et développements spécifiques des faces cristallines, est important pour l’utilisation et l’intégration d’un cristal non-linéaire dans un système optique (ex : prisme droit à faces parallèles ┴ aux directions d’accord de phases).
Les problèmes liés au désordre, auxquels le laboratoire de Cristallographie s’intéresse sont de plusieurs sortes: (i) Accès aux désordres structuraux à partir des diagrammes de diffraction sur poudres. Modélisation du profil observé; (ii) Taux de désordre entre phases ordonnées et désordonnées. Molécules pharmaceutiques mal cristallisées; (iii) Désordres intermoléculaires et défauts de périodicité; (iv) Transitions ordre-désordre; (v) Composés amorphes. Les systèmes étudiés qui présentent un taux de désordre vont des composés inorganiques comme les conducteurs ioniques ou les oxydes supraconducteurs, aux molécules médicaments. Les verres aussi sont étudiés au laboratoire. Il s'agit de verres métalliques préparés par coulée lente qui possèdent de très bonnes qualités mécaniques; ils peuvent donc être travaillés (filage, moulage) dans une large gamme de température sans recristalliser. Des expériences de cristallisation in situ ont été réalisées en diffraction des neutrons afin d'étudier l'influence de la vitesse de chauffage sur ces alliages amorphes.
III.3.2. Matériaux macromoléculaires:
Les méthodes de résolution de structures tridimensionnelles des polymères (synthétiques ou naturels) requièrent la combinaison des méthodes de diffraction (rayons-X, électronique) et des méthodes de simulations numériques. En effet, ces macromolécules ne peuvent pas former des cristaux de taille suffisante pour pouvoir être étudiées par les méthodes classiques de cristallographie. Dans certains cas, il est cependant possible d’obtenir un nombre limité de données de diffraction. En effet, certains polymères existent sous forme cristalline ou semi-cristalline à l’état natif. Il est également parfois possible d’ordonner des polymères sous forme de fibres, de films ou de monocristaux de petites tailles. De tels monocristaux obtenus par croissance en solution possèdent généralement une morphologie de type plaquette, d’une épaisseur de quelques dizaines d’Angströms, et d’une surface de l’ordre du micron2. Si la dimension de ces cristaux ne permet pas l’utilisation des techniques de diffraction des rayons-X, la diffraction des électrons est, par contre, parfaitement appropriée à leur étude structurale. Elle permet de déterminer les paramètres de la maille cristalline, et la symétrie du groupe d'espace. Les intensités des réflexions peuvent être évaluées, mais leur nombre reste encore insuffisant pour permettre une localisation des atomes dans la maille cristalline. La structure ne peut être résolue que par la construction préalable de modèles stéréochimiques permettant l’analyse des conformations permises pour la chaîne macromoléculaire à l’état solide. Ces modèles sont ensuite testés sous forme d’arrangements moléculaires régis par les éléments de symétrie de la maille cristalline, puis optimisés en tenant compte simultanément de l’énergie réticulaire et des données de diffraction. Ces méthodes ont permis la résolution de plus d’une dizaine de structures tridimensionnelles de polysaccharides linéaires. Le nombre et la qualité de ces résolutions structurales permettent de tirer des conclusions importantes quant aux transitions conformationnelles, aux polymorphismes et à l’hydratation de cette famille de macromolécules biologiques. Ces résolutions s’accompagnent également de développements parallèles dans les méthodes de simulation et de prédiction de polymorphisme cristallin et d’arrangements des chaînes de polymères dans des systèmes de basse symétrie.
III.3.3. Modélisation mésoscopique :
Les méthodes traditionnelles de modélisation sont centrées sur une description du système à l'échelle moléculaire (atomistique) ou macroscopique (modèle de continuum). En raison des limitations dues à la puissance de calcul des ordinateurs et de la connaissance théorique générale, la région qualifiée de ‘mésoscale’ entre ces deux extrémités n'a pas été exploitée. Cependant des développements récents dans ce domaine de modélisation ont permis l’essor de méthodes permettant d’aborder cette échelle méso.
Les éléments qui constituent les unités de base de cette échelle méso sont décrits par des unités fondamentales plus grandes que les modèles moléculaires, qui exigent un détail atomique. Un modèle de polymère est décrit par ses unités monomères plutôt que par l’ensemble des atomes qui le constituent. Les méthodes de simulation déterminent la structure, les propriétés et la dynamique de ces modèles. Les échelles de taille et de temps des méthodes de 'mésoscale' dépassent très largement celles des échelles de la simulation moléculaire. Il est possible d’étudier les liquides complexes, les mélanges de polymère, et la structuration des matériaux à l'échelle du manomètre ou du micron.
La taille caractéristique des modèles 'mésoscales' se situe entre l'échelle atomique et l'échelle du continuum, c’est à dire entre 10 et 100 nm. Quelques exemples typiques de cette échelle de grandeur sont : (i) les domaines cristallins dans les polymères semi-cristallins; (ii) les particules de latex ; (iii) les fibres et les composites ; (iv) les aggrégats de détergents.
L’augmentation de l'échelle de taille s’accompagne également d’une augmentation de l’échelle du temps, puisque les processus très rapides dus aux mouvements des atomes ne sont pas inclus dans la description du système. Les exemples d’applications de ces méthodes sont : (i) la formation des micelles ; (ii) les propriétés rhéologiques ; (iii) la miscibilité ; (iv) la température de transition vitreuse ; (v) la morphologie des polymères ; (vi) la diffusion.
La méthode Dissipative Particle Dynamics (DPD) est une méthode de simulation 'mésoscale' d’étude des écoulements dans les fluides complexes, tels que des suspensions, les émulsions et les polymères fondus. Elle fait l’objet d'un nombre croissant d'études théoriques et a été appliquée avec succès à l'étude d'une grande variété de systèmes, comme les écoulements dans des milieux poreux, des suspensions colloïdales, des suspensions polymères et des fluides binaires non-miscibles. L'idée fondamentale de DPD est de décrire le système par différentes particules en interaction soumise aux lois de l'hydrodynamique newtonienne du fluide.
III.4. Recherches des Conformations Bio-Actives
Les systèmes biologiques utilisent principalement deux modes de communication : un mode moléculaire et un mode électrique. Dans le mode moléculaire, le message est véhiculé par des molécules spécifiques d'un système de reconnaissance donné. Ces molécules possèdent une conformation "biologiquement" active dont l'activité est un résultat complexe faisant intervenir de nombreux facteurs. Pour qu'un substrat puisse interagir avec un récepteur et induire ainsi une réponse biologique, plusieurs critères doivent être réunis simultanément : elles concernent les propriétés moléculaires globales permettant au substrat de diffuser dans la biophase et atteindre l'environnement immédiat du récepteur, puis les propriétés structurales responsables de la spécificité, de l'affinité et de la réversibilité de la fixation du substrat sur le récepteur. Dans ces conditions, on observe entre le substrat et le récepteur de nombreuses interactions non liées faisant intervenir effets stéréo-électroniques, forces de van der Waals, effets hydrophobes. La fonction d'une molécule résulte donc d'un ensemble de propriétés physico-chimiques observables, elles-mêmes attribuables à une structure moléculaire particulière. Les propriétés sont une fonction directe de la structure, tandis que la structure peut, dans une certaine mesure, être déduite des propriétés.
La découverte d'une activité biologique intéressante requiert l'examen de la structure de la molécule correspondante. Pour caractériser complètement les dépendances entre cette activité et la structure initiale, l'approche la plus légitime consiste à caractériser l'interaction susbtrat-récepteur. Cette approche n'est envisageable que lorsque le récepteur a pu être identifié, isolé, et sa structure tridimensionnelle résolue. Dans ce contexte, un certain nombre d'algorithmes permettent de calculer des propriétés résultant de la structure tridimensionnelle de ce récepteur, d'identifier le ou les sites d'interactions avec le substrat, à l'aide de programmes tels que GRID. Enfin, l'exploration des modes possibles d'arrimage du substrat dans le site actif du récepteur se fait en utilisant des logiciels appropriés qui utilisent les principes de la mécanique moléculaire, voire de la dynamique moléculaire, en prenant éventuellement en compte les molécules de solvant.
Une autre manière d'aborder le problème est l'approche empirique mise en œuvre par les méthodes de relations structure-activité. En l'absence d'information sur le récepteur, les éléments actifs et inactifs d'un échantillon de molécules proches structuralement sont comparés entre eux afin d'établir les relations et les règles empiriques qui régissent leur activité biologique. Quand ces relations sont exprimées quantitativement, on nomme QSAR (Quantitative Structure-Activity Relationship) les méthodes correspondantes. Elles s'appuient sur une description quantitative des structures et activités et fournissent des relations formulées mathématiquement. Parmi les descripteurs structuraux, on trouve habituellement des propriétés globales telles que le point d'ébullition, le volume moléculaire, la réfractivité molaire ou encore le log P, ainsi que des paramètres mesurables tels que les paramètres stériques et électroniques. A l'instar de ces méthodes, CoMFA (Comparative Molecular Field Analysis) exploite pleinement le fait que la structure moléculaire contienne implicitement toute l'information requise à l'établissement de relations structure-activité. Son originalité réside dans l'utilisation de descripteurs structuraux tridimensionnels, qui permettent de prendre en compte l'influence de la topologie et autorisent l'étude de molécules flexibles. Plusieurs champs peuvent être évalués (champs stérique, électrostatique, lipophilie moléculaire). Les modèles sont constitués d'une région conformationnelle tridimensionnelle au sein de laquelle les variations de champ sont utilisées pour expliquer les variations d'activité. Les perturbations occasionnées par l'introduction d'une molécule supplémentaire permettent la prédiction de son activité biologique. Les méthodes de QSAR sont couramment mises en œuvre, pour la prédiction et le "design" de nouvelles molécules biologiquement actives, mais également dans le cadre d'une meilleure caractérisation de mécanismes biochimiques. Leur succès contribue à leur utilisation dans les domaines aussi divers que la biochimie, biologie, toxicologie, physiologie,…
III.5. Les bases de données
L’exposé des différentes facettes de chimie calculatoire permet de mettre en exergue l’impérieuse nécessité de disposer d’accès interactifs à des bases de données. La majorité des programmes de recherche requiert l’utilisation d’au moins une, sinon de plusieurs bases de données. La nature de ces dernières peut être variable : bases de données réactionnelles, bases de données structurales – Cambridge Structural Data Base, Protein Data Base, bases de données génomiques et protéomiques, bases de données propres à des domaines scientifiques (paramètres de champs de forces, banques structurales de motifs monomères, banques non-redondante de motifs structuraux de biomolécules) ou bases de données factuelles.
IV. L’existant.
IV.1- LEDSS
Le service informatique du LEDSS, qui comprend 3 ingénieurs, s’articule autour de 3 grands pôles : Informatique et Chimie Calculatoire, Micro-informatique et Bureautique, Réseaux
Informatique et Chimie Calculatoire: Ce pôle représente une puissance cumulée de plus de 25 GFLOPS et se compose de 6 serveurs IBM dont 2 SMP, de 10 stations SGI, de 4 stations SUN et 1 serveur Linux. Il fournit les ressources de calcul pour les équipes du LEDSS de :Chimie théorique, Modélisation Moléculaire, Cristallographie, RMN. Le service informatique assure la maîtrise, la sécurité, l’exploitation et la gestion de tous les matériels et logiciels du pôle et fournit le support et l’assistance aux utilisateurs.
Micro-informatique et Bureautique: Ce pôle gère une station de travail serveur et 130 micro-ordinateurs pour les besoins de gestion, d’administration et de bureautique des équipes du LEDSS et de toutes les autres composantes de l’UFR
Réseaux: Ce pôle assure la maîtrise et la sécurité des sous réseaux locaux :Sous réseau Calcul; Sous réseau Bureautique; Sous réseau Administration; Sous réseau Enseignement; Sous réseau Externe. Il assure aussi la maîtrise de la connexion aux réseaux de l’UJF et de RENATER en collaboration avec la direction du CRIP.
2- CERMAV
Le service informatique du Cermav composé de deux ingénieurs a en charge l’administration des moyens informatiques de l’unité regroupant les aspects scientifiques, bureautiques et réseaux de façon à assurer le bon fonctionnement général du laboratoire.
Les aspects scientifiques : Le laboratoire dispose d’un serveur SGI bi-processeurs Origin 200 et 18 stations de travail SGI (O2, Indigo et Indy) et 3 stations Sun pour un espace disque total de près de 100 Go. Il fournit les ressources de calcul pour les équipes de recherches qui utilisent la modélisation moléculaire, la Résonance Magnétique Nucléaire, la cristallographie (Rayons-X; électrons), le traitement d'images, la bio-informatique. Le service informatique assure la maîtrise, la sécurité, l’exploitation et la gestion de tous les matériels et logiciels du pôle et fournit le support et l’assistance aux utilisateurs.
Les aspects bureautiques: Le service informatique gère un parc de 110 micro-ordinateurs plus spécifiquement dédiés aux tâches de gestion, d’administration, de bureautique et d'infographie.
Les aspects réseau: L’ensemble des ordinateurs est relié, au travers de deux sous-réseaux utilisant une topologie ethernet 10 BaseT à 10 Mbits, au réseau Grenet, le réseau universitaire Grenoblois lui-même connecté au réseau régional Aramis, partie intégrante de Renater.
3- Laboratoire de Cristallographie
Quatre personnes (3 ingénieurs et un technicien) constituent le service informatique du laboratoire de Cristallographie qui dispose d’un parc de stations de travail : 1 O2 SGI, 2 Dec Alpha 3000, 2 HP 712 et de 4 XP1000 de Compaq pour une capacité totale de 90 Go et d’un parc de 150 micro-ordinateurs.
4- Département de Pharmacochimie Moléculaire
Le Département de Pharmacochimie moléculaire dispose, sur le plan matériel, de trois stations de modélisation : une Indigo Elan et deux O2 mais ne dispose pas, à l'heure actuelle, de personnel informaticien.
V. Prospective technologique pour les quatre prochaines années.
La montée en puissance de la chimie calculatoire est une évolution majeure dans les programmes de chimie à l'échelle nationale. Dans ce contexte, les projets scientifiques des différentes unités de recherche, ont été expertisés et validés à l'occasion de récents comités d'évaluation. Le développement des activités de recherche en chimie calculatoire sont des priorités affichées pour le programme quadriennal en cours.
En complément des activités de recherche relevant directement de la chimie quantique et de la modélisation, il convient de mentionner les besoins d'interfaces que nécessitent les accès partagés et le traitement des données expérimentales d'origine multiple (Résonance Magnétique Nucléaire (solution haute-résolution; solide), cristallographie (cristaux moléculaires, poudres, fibres, films, protéines,…) et traitement des données mesurées sur les Très Grands Equipements Grenoblois (ILL, ESRF), spectroscopies Infra-Rouge et Raman, spectrométrie de masse, microscopies à champs proches. L'intégration de cet ensemble d'équipements lourds, dans le cadre d'un Plateau Technique ouvert à la communauté des chimistes grenoblois se décline souvent la représentation de la Figure 3 qui illustre le rôle très structurant du projet méso-informatique du pôle de chime calculatoire de l'UJF.
Figure 3 : Le plateau technique du pôle chimie
VI . Les Logiciels
Dans le domaine de la modélisation moléculaire, le coût d'utilisation de logiciels professionnels, généralement spécifiques d'un constructeur, représente une charge financière importante. Dès lors, l'acquisition de telles licences ne peut être dissocié de l'acquisition du matériel. Nous avons donc cherché à préciser les besoins logiciels nécessaires à la réalisation des programmes scientifiques des différentes unités de recherche. Ceux-ci sont regroupés ci-dessous articulés autour des cinq grands axes de recherche pour lesquels nous précisons les laboratoires directement impliqués.
Figure 4 : Inventaire des logiciels
Cet inventaire souligne l'importance des logiciels professionnels que sont Cerius et Insight/Discover de la société MSI et Sybyl de Tripos dont l'utilisation est actuellement incontournable. Ceux-ci fonctionnent spécifiquement dans un environnement SGI.
VII. Formation Pédagogique
Dans le cadre de DEA de Chimie-Physique Moléculaire et Structurale (Ecole Doctorale Chimie Sciences du Vivant) des travaux dirigés sont organisés dans les domaines de chimie quantique, modélisation et graphique moléculaire, afin de donner aux étudiants une solide formation pratique dans le domaine de la chimie calculatoire. Ces volets s'inscrivent en complément et en lien avec les cours théoriques reçus lors du DEA. Cela permet aux étudiants d'aborder la mise en œuvre pratique des divers aspects de la chimie théorique: analyse du problème, modélisation du problème, création de fichiers de données, exploitation des résultats, analyse critique des résultats, appréhension des contraintes machine et de rapport temps et coût machine versus qualité du calcul. Pour ce volet de la formation le programme GAUSSIAN est utilisé; ce programme a l'avantage d'être largement utilisé dans ce domaine de recherche puisqu'il englobe presque tous les aspects de la chimie quantique. De plus, l'utilisation des logiciels de visualisation (par exemple MOLDEN) permet d'appréhender de façon conviviale les résultats obtenus : optimisation de géométrie, orbitales moléculaires, calculs de fréquence,.. A la suite de cette formation, les étudiants acquièrent une compétence pratique des techniques utilisées en recherche ainsi que les bases des capacités d'analyse névessaires pour mener une étude en chimie quantique. Dans le domaine de la modélisation et du graphisme moléculaire, les enseignements pratiques sont donnés autour des suites logicielles SYBYL et CERIUS, ce qui permet aux étudiants de se familiariser au niveau d'applications orientées d'une part vers les sciences du vivant et le domaine des matériaux.
VIII. Choix technologiques et mise en œuvre des équipements
L’ensemble des moyens et des prospectives des principaux acteurs des unités de chimie fait que le regroupement au sein d’un grand Pôle de Calcul pour la Chimie Calculatoire est devenu incontournable pour pouvoir s’équiper de matériel plus puissant que celui existant.
Nous souhaitons porter nos choix sur des solutions qui permettent d’intégrer le mieux possible les parcs existants, dans la mesure où ces solutions correspondent véritablement à des besoins exprimés et identifiés par la communauté des chimistes dans le domaine du calcul scientifique.
Les aspects concernant la visualisation graphique pour lesquels les laboratoires apparaissent déjà équipés et le stockage des données des utilisateurs restent pour leur part déportés au niveau des ordinateurs des différentes entités.
Choix du matériel
L'ensemble des besoins exprimés par la communauté scientifique nous a orienté vers une solution bâtie sur 2 types de machines permettant de répondre à la fois aux besoins de calculs intensifs et aux besoins liés à l'utilisation de ces logiciels professionnels :
Cette configuration bi-constructeur, qui est caractéristique de la Chimie Calculatoire, nous permettra d’accroître l’efficacité des calculs car nous pourrons porter nos codes sources sur le matériel le plus adapté, ce qui n’est pas toujours possible pour des laboratoires mono-constructeurs. La forte complémentarité en terme de matériel entre le LEDSS et le CERMAV permettra de résoudre aisément les problèmes d’exploitation .
Ces machines seront des quadriprocesseurs car nos programmes sont généralement peu parallèlisables et les mesures ont montré qu’au delà de 4 processeurs nous perdions en efficacité.
Implantation du site
Le matériel s’intégrera dans la salle machine du LEDSS qui possède déjà toutes les infrastructures nécessaires à la mise en œuvre de gros matériels mutualisés.
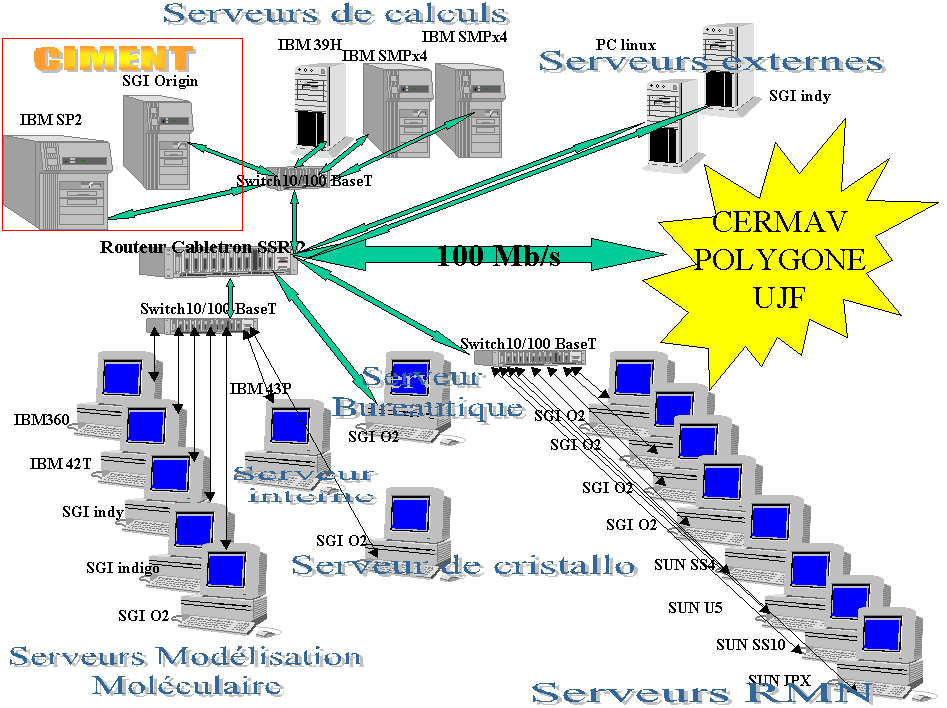
Figure 5 : Intégration des nouveaux matériels dans l’existant
Exploitation du site
Les services informatiques des laboratoires partenaires, se partageront la maîtrise d’œuvre, en utilisant les moyens, les outils et les procédures existant dans les différentes unités de recherche. Les objectifs visés sont d’optimiser l’utilisation équitable des ressources par les différents acteurs et de minimiser les conflits mémoire et CPU. La préservation de l’existant sera la priorité forte de l’exploitation.
La création des données, ainsi que le stockage et la visualisation resteront à la charge de chacun des participants.
Administration système et réseau
Cette charge sera importante, car en plus de l’administration de l’existant, devront être pris en compte les nouveaux matériels, tout en conservant le haut degré de fiabilité et de disponibilité des machines et réseaux existants; les logiciels permettant la centralisation et l’administration de tous les matériels et réseaux seront utilisés.
Formation, support et assistance
Une des missions du service concerne la formation, le support et l’assistance des utilisateurs. Cette tâche qui est déjà assurée dans certains des laboratoires partenaires du projet, revêtira une importance encore plus grande, car c’est d’elle que dépendra le succès des solutions mises en œuvre, et l’intégration des outils de chimie informatique dans les thématiques émergentes issues de l’ensemble des laboratoires de la communauté des chimistes de l’UJF. Il est évident que dans le contexte de partage de ressources, cette importance sera encore accrue, et tout sera mis en œuvre avec les moyens existants pour que l’excellence des services soit préservée.
Interface avec les moyens nationaux
L’utilisation des moyens nationaux (CINES, CIRCE) sera préservée et favorisée par la possibilité de tester les travaux localement avant de les exécuter sur les grands calculateurs nationaux, permettant de décharger d’autant les réseaux. Une deuxième voie d’action consistera à mettre en œuvre des environnements de développements compatibles, des cross-compilateurs,…
IX. Le suivi du programme.
Le suivi du programme sera assuré par un conseil scientifique et de gestion comprenant des membres des différentes Unités de recherche contractantes (directeurs, représentants Ingénieurs, représentants utilisateurs), des représentant de l'UFR de Chimie ainsi que les représentants des différents projets de méso-informatiques du site grenoblois.
Une des missions de ce conseil consisterait à proposer des solutions d'intégration pour des laboratoires ou des thématiques émergentes.
X. Devis
Pour la réalisation de ces projets ont été contactés uniquement les constructeurs de matériels qui correspondaient aux choix scientifiques et aux contraintes logicielles présentées ci-dessus.
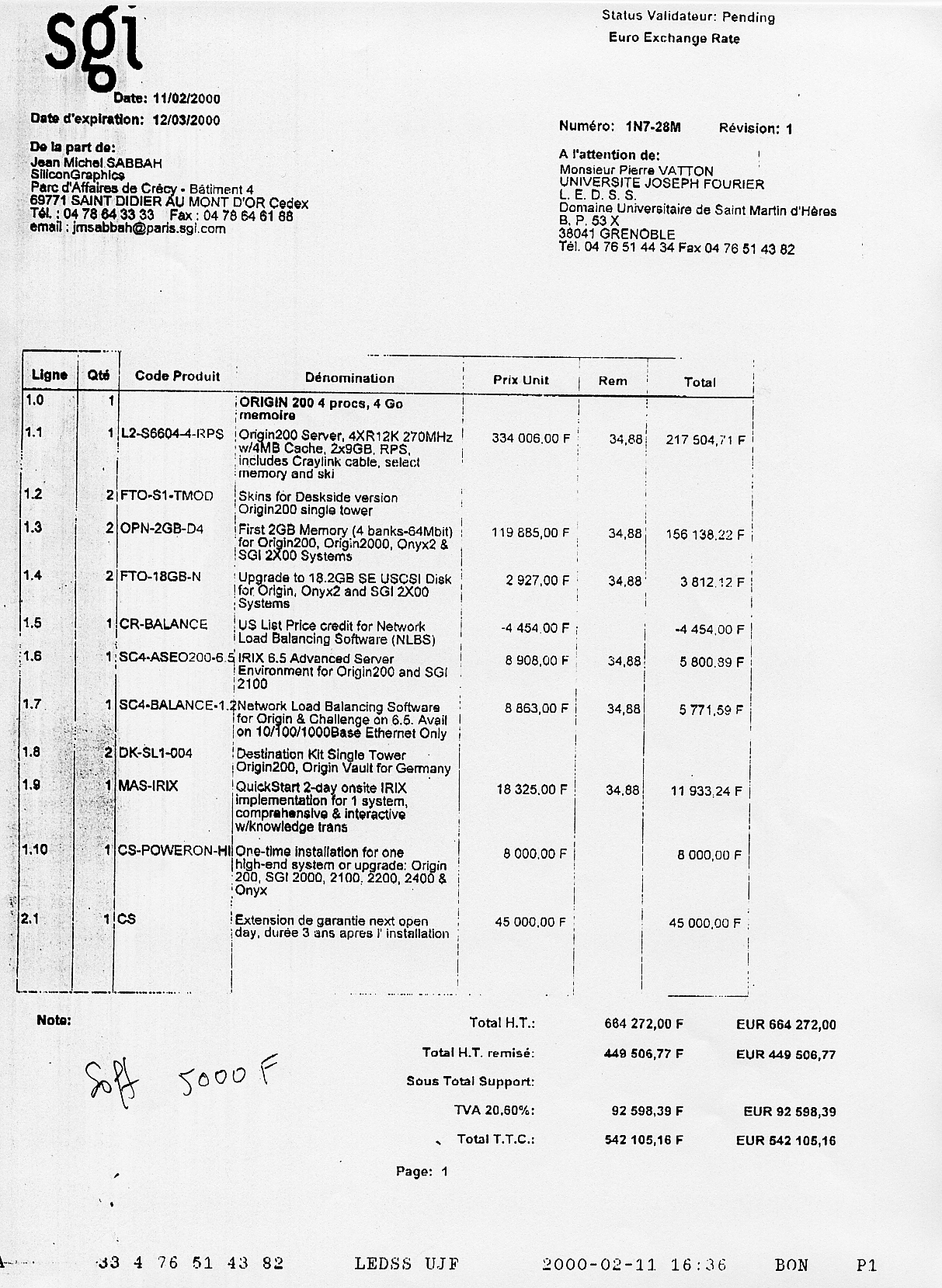
Devis de SGI
Devis d'IBM
XI. Budget prévisionnel et financement.
Investissement.
L'estimation a été réalisée en partant sur une configuration de 2 machines et en incluant une partie correspondant à un certain nombre de logiciels dont l'accès serait réservé en priorité aux chercheurs et enseignants-chercheurs appartenant à des unités qui ne possèdent actuellement pas de ressources matérielles et/ou logicielles mais qui feraient des demandes de calculs.
IBM: 1400 KF
SGI: 500 KF
Bibliothèque de modules de modélisation : 100 KF
Total : 2000 KF
Fonctionnement.
Les contrats de maintenance des machines étant payés à l'achat, ils n'entrainent pas de frais de fonctionnement, pendant les trois premières années. Il faut par contre prévoir, pour ces contrats de maintenance, une participation à une hauteur d'environ 200-300 KF par an à partir de l'année 3 du début du programme.